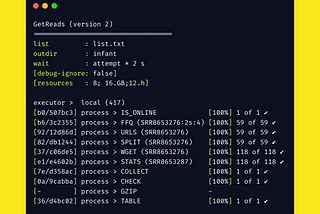
Andrea Telatinin#!/ngs/shDownloading NGS datasets using NextflowA simple to use pipeline to download FASTQ files from NCBI or EBI, which serves as a good advertisment for my favourite workflow managerJul 29, 2022Jul 29, 2022
Andrea Telatinin#!/ngs/shManage shared software with environment “modules”How to setup “module load” in your shared Linux serverJun 12, 2020Jun 12, 2020
Andrea TelatinStoring command line Git credentialsA short note on avoiding typing your password at every pushApr 16, 2019Apr 16, 2019
Andrea Telatinin#!/ngs/shCompressing FASTQ files for long term storageThe raw output of NGS experiments should be backed, and format-specific compressor like “dsrc2” comes handy for thisFeb 7, 2019Feb 7, 2019
Andrea Telatinin#!/ngs/shA simple introduction to XML and JSONTwo popular formats to store structured dataNov 27, 2018Nov 27, 2018
Andrea Telatinin#!/ngs/shMerging Illumina lanes with a Bash scriptThis article describes a Bash script to perform a simple task with some controls and best practices.Oct 15, 2018Oct 15, 2018
Andrea Telatinin#!/ngs/shPigz: a faster alternative to gzip for big filesUsing multiple threads to compress files but maintaining compatibility with gunzipOct 2, 20181Oct 2, 20181
Andrea Telatinin#!/ngs/shIs a Bash variable set?Preventing unbound variables is good, but we need a way to tell if a variable has no content or has never been initializedSep 27, 2018Sep 27, 2018
Andrea Telatinin#!/ngs/shA short introduction to Git and GitHubThe minimum workflow for keeping file revisions under controlJun 27, 2018Jun 27, 2018